
Na rysunku powyżej widać fragment programu do wyszukiwania dużych delecji w wynikach z 23andMe. Wyniki z 23andMe nie są stworzone do tego aby szukać w nich delecji. Jeśli szukasz dużych delecji (~100 tyś. bp) to dobrym sposobem wydają się mikromacierze, jeśli szukasz krótkich delecji (~100 bp) to badania WES/WGS będą pomocne. Do wyniku wyszukiwania dużych delecji w wynikach z 23andMe należy podchodzić ostrożnie. Wyszukiwanie opiera się na kilku założeniach. Po pierwsze zakładam, że delecja na dwóch bliźniaczych chromosomach skutkuje wartością '--' w wyniku. Taka wartość może się też pojawić, niestety z innych powodów. Druga sprawa to taka, że w dużym fragmencie kolejnych wariantów z wartością '--' nie dopuszczam innych wartości, żeby uznać dany fragment za delecję. Takie pojedyncze warianty z innymi oznaczeniami mogłyby się pojawić za sprawą homologów/ortologów/paralogów czy pseudogenów. Jeśli tak będzie to taki fragment nie zostanie uznany za delecję (być może przydałoby się wprowadzić do programu pewną tolerancję na tego typu rzeczy). O ile delecja na parze chromosomów wydaje się, że skutkuje oznaczeniem '--' (w wynikach są też oznaczenia 'DD' oznaczające delecje, ale nie o takie delecje chodzi) to delecja na pojedynczym chromosomie jest bardziej problematyczna. Prawdopodobnie skutkuje ona tym, że pewien spory fragment kolejnych wyników dla wariantów składa się wyłącznie z genotypów o dwóch takich samych allelach. Ta sytuacja wcale nie musi być patologiczna i nie musi oznaczać delecji. Żeby błędnie nie zakwalifikować takiego czegoś jako delecji należało ustawić duże wartości w parametrach dla wykrywania delecji na pojedynczym chromosomie.
Obsługa programu jest bardzo prosta. Po naciśnięciu przycisku "przeglądaj" wybiera się plik z surowymi danymi. Program rozpoczyna pracę automatycznie najpierw pobierając plik, następnie go odczytując. W kolejnej fazie zaznacza na obrazku z chromosomami odpowiednie obszary, które podlegały genotypowaniu, na podstawie wczytanego pliku. W tym samym czasie próbuje wykryć delecje i jeśli tak się stanie rysuje odpowiednie informacje i dodaje info o tym co zostało znalezione (na spodzie strony). Oznaczone w wyniku warianty rysowane są kolorem zielonym, znalezione delecje na obu chromosomach kolorem czerwonym a na jednym chromosomie kolorem pomarańczowym. Można zmienić parametry wyszukiwania i/lub skalę obrazka i nacisnąć przycisk rysuj. Wykres zostanie ponownie narysowany dla zmienionych danych.
Jakie delecje można wykryć
Wykrycie delecji zależy od gęstości wariantów w danym zakresie i od ustawień programu. Jeśli ustawimy, że interesują nas duże delecje to jeśli program coś wykryje to jest większa szansa, że to co zrobił jest poprawne. Jeśli zmniejszymy parametry programu tak, żeby wykrywać mniejsze delecje to program stanie się bardziej "czuły", ale zaczynają wkradać się nieprawidłowości (wyniki fałszywie pozytywne). Poniżej przedstawionych zostało kilka przykładów wyszukiwania delecji w (spreparowanych) wynikach z 23andMe. Przykłady zostały przygotowane na podstawie dostępnej literatury medycznej opisującej wykryte delecje.
Delecja 22q11

Na rysunku obok widzimy delecję na 22 chromosomie w okolicach locus q12. Obrazek przedstawie delecje dla trzech różnych pacjentów powiększone w programie do wielkości x8. Kolorem czerwonym zaznaczono co prezentowałby program gdyby delecja dotyczyła obu chromosomów, kolorem pomarańczowym gdyby delecja dotyczyła tylko jednego chromosomu. Delecja tego regionu występuje z częstotliwością 1/4000 i skutkuje opóźnieniem rozwoju, opóźnieniem mowy, problemami z nauką, zachowaniami ze spektrum autyzmu i ADHD. Czasami symptomy mogą być na tyle łagodne, że opóźniają właściwą diagnozę.
Delecja 22q13

Delecja 22q13.3 nosi też nazwę zespołu Phelan-McDermida. Częstotliwość występowania nie jest znana, ale podejrzewa się, że jest bardzo niska. W rejonie 22q13 występuje gen SHANK3, silnie powiązany z autyzmem. Dlatego też delecje w rejonie 22q13 skutkują objawami ze spektrum autyzmu. Na rysunku obok widać rzeczywistych 5 przypadków delecji w tym rejonie zaczerpniętych z literatury. To co się rzuca w oczy to to, że rozmiary tych delecji są radykalnie różne. W przypadku drugiego pacjenta widać, że doszło do delecji całego locus q13.31, podczas gdy u trzeciego pacjenta widać, że brakuje tylko krótkiego fragmentu w locus q13.33. Co więcej, ta delecja nie zostanie znaleziona przez opisywany program na standardowych ustawieniach, trzeba zmniejszyć do 10 ilość kolejnych wariantów z '--' w parametryzacji.
Delecja 1p36
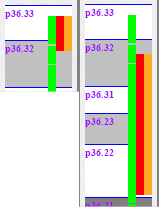
Po lewej widzimy delecję na pierwszym chromosomie. Występuje z częstotliwością około 1/5000. Osoby z tą delecją dotknięte są często różnymi dymorfizmami. Ich rozwój umysłowy jest także nieprawidłowy.
Konkluzja
Wiele osób wykonuje badania w 23andMe. Nie są to drogie badania, ale nie niosą tyle informacji co np.: WES czy NGS. Wydaje się, że dziecko dotknięte różnymi dymorfizmami, cierpiące na padaczkę czy z ciężkim upośledzeniem umysłowym powinno mieć pogłębioną diagnostykę genetyczną. W praktyce okazuje się, że badania kariotypu czy MLPA niczego nie wykrywają a z uwagi na to, że za mikromacierze czy WES muszą zapłacić rodzice to badania te nie są wykonywane. Opisywany tutaj program stara się wykryć duże delecje w wynikach z 23andMe. Oczywiście, wszystkiego co wykryje ten program nie należy brać za "dobrą" monetę i trzeba potwierdzić znalezisko w odpowiednim badaniu genetycznym (mikromacierze, WES). Być może istnieją osoby, które nie zdecydowały się na mikromacierze czy WES a wykonały badanie w 23andMe i może są rzeczywiście posiadaczami jakiejś dużej delecji. Opisywane na tej stronie przykłady kilku delecji nie wyczerpują możliwości opisywanego programu. Stanowią jedynie przykład. Jeśli ktoś ma ochotę to zapraszam do korzystania z programu. Należy się wyczulić na to, że program prawdopodobnie pokaże problem w rejonie chromosomów X i/lub Y. Dla kobiet będzie zgłaszał błąd, że nie ma dużej ilości danych na chromosomie Y (hmmm, właściwie całego chromosomu). W przypadku chromosomu X wydaje się, że 23andMe ma jakiś problem z genotypowaniem i czasem brakuje dużych fragmentów tego genu. Nie sądzę, że tak jest w rzeczywistości, raczej to błąd po stronie 23andMe. Wydaje się, że można spokojnie zignorować wskazywane delecje na chromosomie X i Y.
Program znajduje się pod adresem: http://agdziedrugijez.pl/hg19/cytobands/cytobands.html







